Синдром Хантера
Синдром Хантера — досить рідко зустрічається генетична хвороба, що вражає тільки представників чоловічої статі, характеризується недостатньою кількістю лизосомального ферменту, внаслідок чого довгі ланцюжки молекул цукру (мукополісахариди) не руйнуються правильно і починають накопичуватися в клітинах організму.
Види
залежно від ступеня вираженості симптоматики та різновиди генної мутації виділяють три форми синдрому Хантера:
- легка — дозволяє вести нормальну життєдіяльність, реалізовувати розумовий потенціал у професійній галузі, в більшості випадків діти таких людей народжуються здоровими;
- среднетяжелая — ознаки з'являються найчастіше у віці від 3 до 8 років (в окремих випадках до 13), інтелект зберігається, тривалість життя може варіюватися від 50 до 60 років;
- важка — прояв симптомів починається в більш ранньому віці (в період від 18 до 36 місяців), зачеплені багато органів і системи, швидке прогресування, помітна розумова відсталість, летальний результат наступає протягом 10-15 років.
Причини
Захворювання передається спадковим шляхом. Організм людини, що страждає даними відхиленням, не може виробляти певний тип білка, що відбивається в його послідовності ДНК. Щоб хвороба проявилася у дівчинки, необхідно щоб вона отримала дефектний ген від обох батьків. За всю історію медицини були описані всього два таких випадки. Зазвичай представниці жіночої статі можуть бути тільки носіями подібного гена.
Симптоми
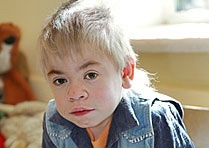
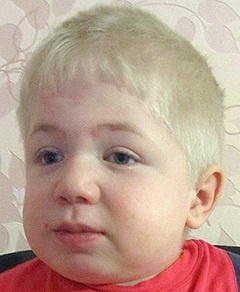
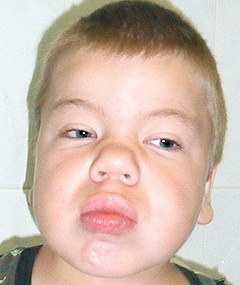
Спектр ознак, який притаманний даному відхиленню, досить широкий. У нього входять як дефекти зовнішності, так і відхилення в розумовому розвитку, працездатності різних органів і систем. Найбільш часто зустрічаються:
- відсталість в розумовому розвитку різного ступеня;
- м'язова слабкість;
- проблеми з розмовною мовою;
- інфекційні захворювання вух, глухота;
- труднощі при пересуваннях;
- відхилення в роботі серцевих клапанів;
- товста груба шкіра;
- голос хриплуватий і низький;
- аномальна потовщення кісток;
- короткі руки з товстими викривленими пальцями;
- збільшені розміри селезінки і печінки;
- погана здатність до навчання в дитячі роки;
- коротка шия;
- рідкіснізуби;
- голова, губи і язик великого розміру;
- сповільненість розвитку і зростання;
- вузлові висипання на шкірі плечей, верхньої частини спини, стегон;
- коліки, оніміння і слабкість у верхніх кінцівках;
- агресивність;
- гучне дихання;
- помутніння рогівки;
- поява в області попереку "монгольських плям";
- підвищена активність;
- тунельний синдром зап'ястя;
- швидке зростання волосся, густі брови;
- хронічна діарея, інші проблеми з кишечником;
- грубі риси обличчя;
- труднощі з дихальноюактивністю, з паузами в диханні вночі, наявність апное сну;
- іноді присутні судоми.
Досить часто супутніми захворюваннями є: часті простудні хвороби, ГРВІ, грип, ангіна, гіпертрихоз, риніт, дизостоз, отит, трахеїт, ларингіт, пневмонія, пупкова і пахова грижі, кіфосколіоз, водянка яєчка, остеоартрит, гідроцефалія, кардіоміопатія, спастична параплегія, атиповий пігментний ретиніт і інші.
Діагностика
Первинний діагноз може поставити педіатр, але для підтвердження необхідно записатися на прийом до лікаря-генетика.
Виявити патологію і визначити ступінь відхилень в різних системах організму, крім клінічних ознак, допомагає проведення таких досліджень:
- аналізи сечі і крові;
- виявлення генеалогічних схильностей ;
- молекулярно-генетичні тести;
- рентгенографія хребта, черепа і трубчастих кісток;
- електрокардіограма;
- ЕхоКГ;
- ультразвукова діагностика органів, розташованих в черевній порожнині;
- магнітно-резонансна томографія головного мозку;
- біопсія тканин міокарда, печінки і шкіри.
Під час вагітності жінки можуть проводити внутрішньоутробну пренатальну перевірку для своєчасного виявлення захворювання.
Лікування
Така хвороба вимагає постійного і комплексного лікування. Тому знадобляться консультації лікаря-кардіолога, окуліста, отоларинголога, логопеда, психолога, психіатра, дефектолога, пульмонолога, ортопеда, невролога. Пацієнтам необхідна довічна ферментозаместітельной терапія. У світі на даний момент зареєстрований тільки один препарат для прийому людям з синдромом Хантера — Елапраза (ідурсульфаза).
Також здійснюється підтримує оздоровлення за допомогою:
- лікувальної фізкультури;
- фізіотерапевтичних процедур (магнітотерапія, електрофорез, лазеропунктура, парафінотерапія);
- вживання вітамінного комплексу, гепатопротекторів, антиоксидантів, цітопротекторов.
На ранніх етапах виявлення патологічного стану послабити симптоматику може пересадка кісткового мозку.
Профілактика
У повсякденному житті потрібно постаратися з дитинства максимально наблизити хворого до нормального повноцінного спілкування з іншими людьми, отримання інформації, яку він може засвоїти, розвитку навичок і умінь. Незважаючи на слабкість здоров'я, люди з легкою формою захворювання можуть стати повноцінним учасником товариства, заводити сім'ю і дітей, працювати.