Мукополісахарідоз
Мукополісахарідоз — це загальна назва досить рідко зустрічаються генних захворювань. Вони безпосередньо пов'язані з патологією обміну кислих глікозаміногліканів, які, в свою чергу, були викликані дефіцитним кількістю лізосомних ферментів обміну глікозаміногліканів.
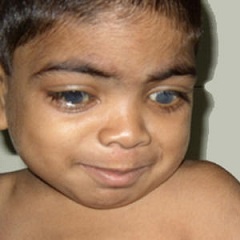

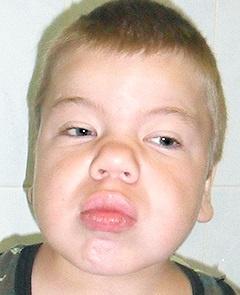
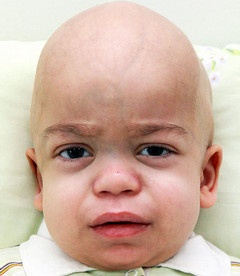
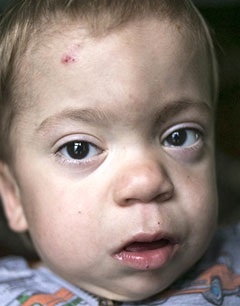
Види захворювання
За характером ферментативного дефекту (виражених кісткових і обмінних змінах і психічних патологій) мукополісахарідоз буває декількох основних типів:
- I тип, який має три основних фенотипу — це синдром Гурлер, синдром Гурлера-Шейе, синдром Шейе, які характеризуютьсянедоліком альфа-L-ідуронідазов, швидким розвитком, наявністю великої кількості В і гепарітінсульфата в сечі;
- II тип — синдром Хантера, для якого характерні пігментний ретиніт, туговухість, велика кількість гепарітінсульфата і хондроітінсульфата В в сечі;
- III тип — синдром Санфіліппо, для якого характерні важке слабоумство і наявність великої кількості гепарітінсульфата в сечі;
- IV тип — синдром Морков, для якого характерні сильна деформація скелета, відсутність помутніння рогівки, відсутність зниження інтелекту і появи грубих рисособи;
- V тип — синдром Шейе, для якого характерні помутніння рогівки і помірно-виражене викривлення скелета;
- VI тип — синдром Марото — Ламі, для якого характерні уповільнення в зростанні, бочкообразная грудна клітка, грубі риси обличчя;
- VII тип — синдром Слая дефіцит р-глюкуронідази, для якого характерний недолік бета-глюкуронідази.
Всі ці типи мають несприятливий прогноз для хворих.
Чим пацієнт стає старше, дорослішає, тим більші зміни скелета у нього трапляються, тим більше спостерігається патологій в різних системах і органах.
Причини мукополисахаридоза
Основна причина появи у людини хвороби мукополисахаридоза пов'язана зі спадковими аномаліями обміну, тобто, з успадкуванням цієї хвороби по аутосомно-рецесивним типом.